Complaint Reporting (AI-Powered Analysis)
The Complaint Reporting module enhances regulatory compliance workflows by deploying AI-powered analysis to determine whether a field event needs to be formally reported to regulatory bodies (like the FDA or EMA). It ensures that critical device malfunctions and patient harms are accurately evaluated against complex legal thresholds.
Step 1: Identifying the Product Sub-Category
The initial phase of a regulatory assessment demands an exact clarification of the product involved, as different devices adhere to distinct reporting guidelines.
- In the Product Information block, access the Product Type field.
- Select the specific device category (e.g., Dental implants, Bladder stimulators, or Infusion pumps). Expert insight: Identifying the precise device classification is vital because the AI uses this context to reference specific risk profiles and historical precedent within its underlying regulatory database.
Step 2: Logging the Adverse Event
Document the precise nature of the failure or the clinical injury sustained during the device's operational use.
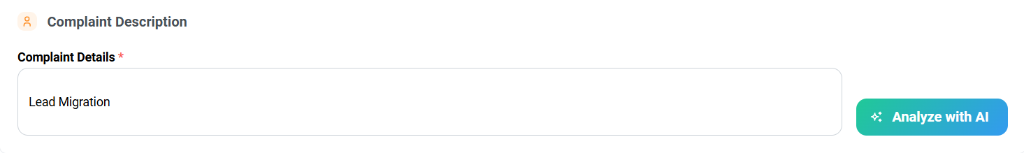
- Proceed to the Complaint Description section.
- Under Complaint Details, enter the event synopsis (e.g., "Lead Migration"). It is recommended to include both the technical failure and the resultant patient condition if applicable.
- Click the Analyze with AI button. The system initiates a semantic evaluation of your input against global medical device reporting regulations.
Step 3: Assessing AI Regulatory Determinations
Upon completion of the analysis, the system renders an instantaneous regulatory verdict accompanied by a detailed clinical rationale.
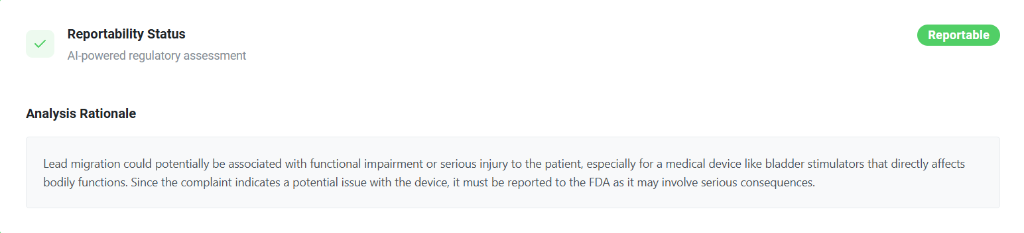
Reportability Status
The platform outputs a definitive, color-coded status indicator:
- Reportable (Green/Flagged): The system has determined the event meets the criteria for mandatory regulatory submission (e.g., MDR to the FDA).
- Non-Reportable (Gray): The event does not cross the threshold requiring agency notification.
Analysis Rationale
To ensure absolute transparency and auditability, the system provides a comprehensive Analysis Rationale. For example, if "Lead Migration" is reported for a Bladder stimulator, the AI explains: "Lead migration could potentially be associated with functional impairment or serious injury to the patient... Since the complaint indicates a potential issue with the device, it must be reported to the FDA as it may involve serious consequences."
This generated rationale can be directly incorporated into official Medical Device Reporting (MDR) documentation, establishing a solid foundation of clinical justification for auditors and regulatory inspectors.